
![]()
![]()
![]()
TBSS repository is developed by Tashrif Billah, Sylvain Bouix, and Ofer Pasternak, Brigham and Women’s Hospital (Harvard Medical School).
If this repository is useful in your research, please cite as below:
Billah, Tashrif; Bouix, Sylvain; Pasternak, Ofer; Generalized Tract Based Spatial Statistics (TBSS) pipeline, https://github.com/pnlbwh/tbss, 2019, DOI: https://doi.org/10.5281/zenodo.2662497
See installation instruction here.
Table of Contents
- Usage
- Useful commands
- Overview
- Branches/Templates
- Caselist
- Input images
- Space
- List of outputs
- multi-modality TBSS
- List creation
- Analysis
- QC
- Fill holes
- Resource profile
- Multi threading
- NRRD support
- Troubleshooting
- Reference
Table of Contents created by gh-md-toc
Usage
usage: tbss_all [-h] [--modality MODALITY] [-i INPUT] [--generate]
[-c CASELIST] [-o OUTDIR] [--studyTemplate] [--enigma]
[--fmrib] [--template TEMPLATE] [--templateMask TEMPLATEMASK]
[--skeleton SKELETON] [--skeletonMask SKELETONMASK]
[--skeletonMaskDst SKELETONMASKDST] [-s SPACE] [-l LABELMAP]
[--lut LUT] [--qc] [--avg] [--force] [--verbose] [-n NCPU]
[--SKEL_THRESH SKEL_THRESH]
[--SEARCH_RULE_MASK SEARCH_RULE_MASK] [--status]
TBSS at PNL encapsulating different protocols i.e FSL, ENIGMA, ANTs template etc.
optional arguments:
-h, --help show this help message and exit
--modality MODALITY
Modality={FA,MD,AD,RD...} of images to run TBSS on
(i) single modality analysis: you must run --modality FA
first, then you can run for other modalities such as
--modality AD
(ii) multi modality analysis: first
modality must be FA, and then the rest i.e --modality
FA,MD,AD,RD,... files from FA TBSS analysis are used
in rest of the modalities
-i INPUT, --input INPUT
(i) DWI images and masks: a txt/csv file with
dwi1,mask1\ndwi2,mask2\n... ; TBSS will start by
creating FA, MD, AD, and RD; additionally, use
--generate flag
(ii) single modality analysis: a
directory with one particular
Modality={FA,MD,AD,RD,...} images, or a txt/csv file
with ModImg1\nModImg2\n... TBSS will be done for
specified Modality
(iii) multi modality analysis:
comma-separated multiple input directories
corresponding to the sequence of --modality, or a
txt/csv file with
Mod1_Img1,Mod2_Img1,...\nMod1_Img2,Mod2_Img2,...\n...
; TBSS will be done for FA first, and then for other
modalities.
(iv) separate nonFA TBSS: if you wish to
run TBSS for other modalities in future, files created
during FA TBSS will be integrated into the nonFA TBSS.
Provide the same --outDir as that of FA TBSS.
All other directories will be realized relative to --outDir.
On the other hand, provide --input and --modality in the same way
as you would provide for FA TBSS.
--generate generate diffusion measures for dwi1,mask1\n... list
-c CASELIST, --caselist CASELIST
caselist.txt where each line is a subject ID
-o OUTDIR, --outDir OUTDIR
where all outputs are saved in an organized manner,
for separate/future nonFA TBSS--it must be the same as
that of previous FA TBSS where all outputs are saved in an organized manner
--studyTemplate create all of template, templateMask, skeleton, skeletonMask, and skeletonMaskDst
--enigma use ENGIMA provided template, templateMask, skeleton,
skeletonMask, and skeletonMaskDst, do JHU white matter
atlas based ROI analysis using ENIGMA look up table
--fmrib use FSL provided template, and skeleton
--template TEMPLATE
an FA image template (i.e ENIGMA, IIT), if not
specified, ANTs template will be created from provided
images, for ANTs template creation, you must provide
FA images, once ANTs template is created, you can run
TBSS on non FA images using that template
--templateMask TEMPLATEMASK
mask of the FA template, if not provided, one will be created
--skeleton SKELETON
skeleton of the FA template, if not provided, one will be created
--skeletonMask SKELETONMASK
mask of the provided skeleton
--skeletonMaskDst SKELETONMASKDST
skeleton mask distance map
-s SPACE, --space SPACE
you may register your template (including ANTs) to
another standard space i.e MNI, not recommended for a
template that is already in MNI space (i.e ENIGMA,
IIT)
-l LABELMAP, --labelMap LABELMAP
labelMap (atlas) in standard space (i.e any
WhiteMatter atlas from ~/fsl/data/atlases/
--lut LUT look up table for specified labelMap (atlas)
--qc halt TBSS pipeline to let the user observe quality of registration
--avg average Left/Right components of tracts in the atlas
--force overwrite existing directory/file
--verbose print everything to STDOUT
-n NCPU, --ncpu NCPU
number of processes/threads to use (-1 for all
available, may slow down your system, default 4)
--SKEL_THRESH SKEL_THRESH
threshold for masking skeleton and projecting FA image
upon the skeleton, default 0.2
--SEARCH_RULE_MASK SEARCH_RULE_MASK
search rule mask for nonFA TBSS, see "tbss_skeleton
--help", default /home/tb571/fsl/data/standard/LowerCingulum_1mm.nii.gz
--status prints progress of TBSS pipeline so far
--noFillHole do not fill holes inside the brain in diffusion measure
--noAllSkeleton do not merge skeletons
--noHtml do not generate summary.html file containing screenshots of axial, lateral, and sagital views
Useful commands
1. Run ENIGMA TBSS
See details on ENIGMA TBSS.
lib/tbss_all -i IMAGELIST.csv \
-c CASELIST.txt \
--modality FA,MD,AD,RD
--enigma \
-o ~/enigmaTemplateOutput/
IMAGELIST.csv is a list of FA,MD,AD,RD images in separate columns. A particular diffusivity images for all cases need
NOT to be in the same directory. Rather, they can be anywhere in your machine. Just make sure to specify absolute
path to the diffusivity image in designated column of IMAGELIST.csv. See details in ENIGMA branch.
NOTE For multi-modality TBSS like above, make sure to have FA as the first modality.
2. Run user template based TBSS
lib/tbss_all -i FAimageDIR,MDimageDIR \
--modality FA,MD \
-c CASELIST.txt \
--template your_FA.nii.gz \
--skeleton your_skeleton_FA.nii.gz \
-o ~/userTemplateOutput/
Alternative to the IMAGELIST.csv in the above, you can specify a directory corresponding to each modality you want to analyze.
However, you have to copy your diffusivity images in a directory. In the very least, the images across FAimageDIR,MDimageDIR
should have caseID from CASELIST.txt somewhere in their file names. But it doesn’t need to have keyword “FA/MD” in their file names.
On the other hand, your_FA.nii.gz and your_skeleton_FA.nii.gz are the templates you should provide.
See details in User template branch.
3. nonFA TBSS
The best way to perform nonFA TBSS is running together with FA such as --modality FA,MD,AD --input faDir,mdDir,adDir.
However, if you want to run nonFA TBSS at a future date based on FA template/registration, you can do as follows:
nonFA --engima TBSS
$libDir/tbss_all -i MD/origdata,RD/origdata \
-o $testDir/enigmaTemplateOutput/ \ # transform files are obtained from here
--modality MD,RD --enigma # --enigma tells to use enigma templates
nonFA --studyTemplate TBSS
$libDir/tbss_all -i AD/origdata \
-o $testDir/studyTemplateOutput/ \
--modality AD \
--studyTemplate # --studyTemplate tells to use previous templates from studyTemplateOutput/stats/*
A few other arguments are obtained by parsing previous command stored in *TemplateOutput/log/commands.txt file. In a nut-shell,
nonFA TBSS would run under the same settings of FA TBSS.
4. Minimum TBSS command
lib/tbss_all -i FAimageDIR \
-c CASELIST.txt \
-o ~/fmribuserTemplateOutput
Voila! The pipeline will create a study specific template. Default --modality is assumed to be FA. See details in
study template branch.
5. ROI analysis
With all the above, you may provide an atlas and a space of the atlas defining image. Then, ROI based statistics will be calculated.
--labelMap JHU-ICBM-labels-1mm.nii.gz \
--lut data/ENIGMA_look_up_table.txt \
--space $FSLDIR/data/standard/FMRIB58_FA_1mm.nii.gz
Even better, ENGIMA branch does ROI based analysis as default.
6. Check progress
If you have a good number of cases to process, and you would like to know how far the pipeline has progressed, do the following:
lib/tbss_all --status --outDir ~/userTemplateOutput/
The --status command uses information from outDir/log/config.ini to collect information about the ongoing TBSS study. It will print a dashboard like below:
Output directory: ~/my_output_directory
Number of cases to process: 228
Progress of FA TBSS:
origdata obtained: 228
pre-processed: 228
registered to template space: 228
skeletonized: 228
roi-based stat calculated: 228
Time taken so far: 2 days, 17 hours, 54 minutes and 3 seconds
Amazing, isn’t it!
7. Create summary
Finally, TBSS pipeline can generate an HTML file with skeleton overlaid upon the diffusivity measure for all cases. As of recent enhancements, HTML file is generated by default. Yet, see the following examples for generating it separately:
lib/writeHtml.py --dir tbss/output/directory --modality MD --ncpu 8
lib/writeHtml.py --dir tbss/output/directory --cut_coords enigma # default --modality is FA
lib/writeHtml.py --dir tbss/output/directory --cut_coords fmrib # default --modality is FA
lib/writeHtml.py --dir tbss/output/directory --modality FW --cut_coords 1,-19,14 (comma separated, no spaces)
The default --cut_coords is auto which resolves to the method find_xyz_cut_coords(img, mask_img=None, activation_threshold=None)
defined in nilearn.plotting.find_cuts.
However, we experimentally devised --cut_coords enigma == 1,-19,14 and --cut_coords fmrib== -17,-24,14 for fixing the cut coordinates
to be used across all images in a study.
Overview
This is Generalized Tract Based Spatial Statistics (TBSS) pipeline,
encompassing different protocols such as ENIGMA
and FSL. It is elegantly designed so you no longer have to
deal with naming your folders/files according to a protocol. It uses some command line tools relevant to
skeleton creation from FSL while replacing all FSL (i.e flirt, applyWarp etc) registration steps by ANTs.
In a nutshell, this pipeline should facilitate an user in running TBSS study by giving more liberty with inputs.
Moreover, it harnesses multiprocessing capability from Python making the program significantly faster than any
job scheduling framework (i.e lsf).
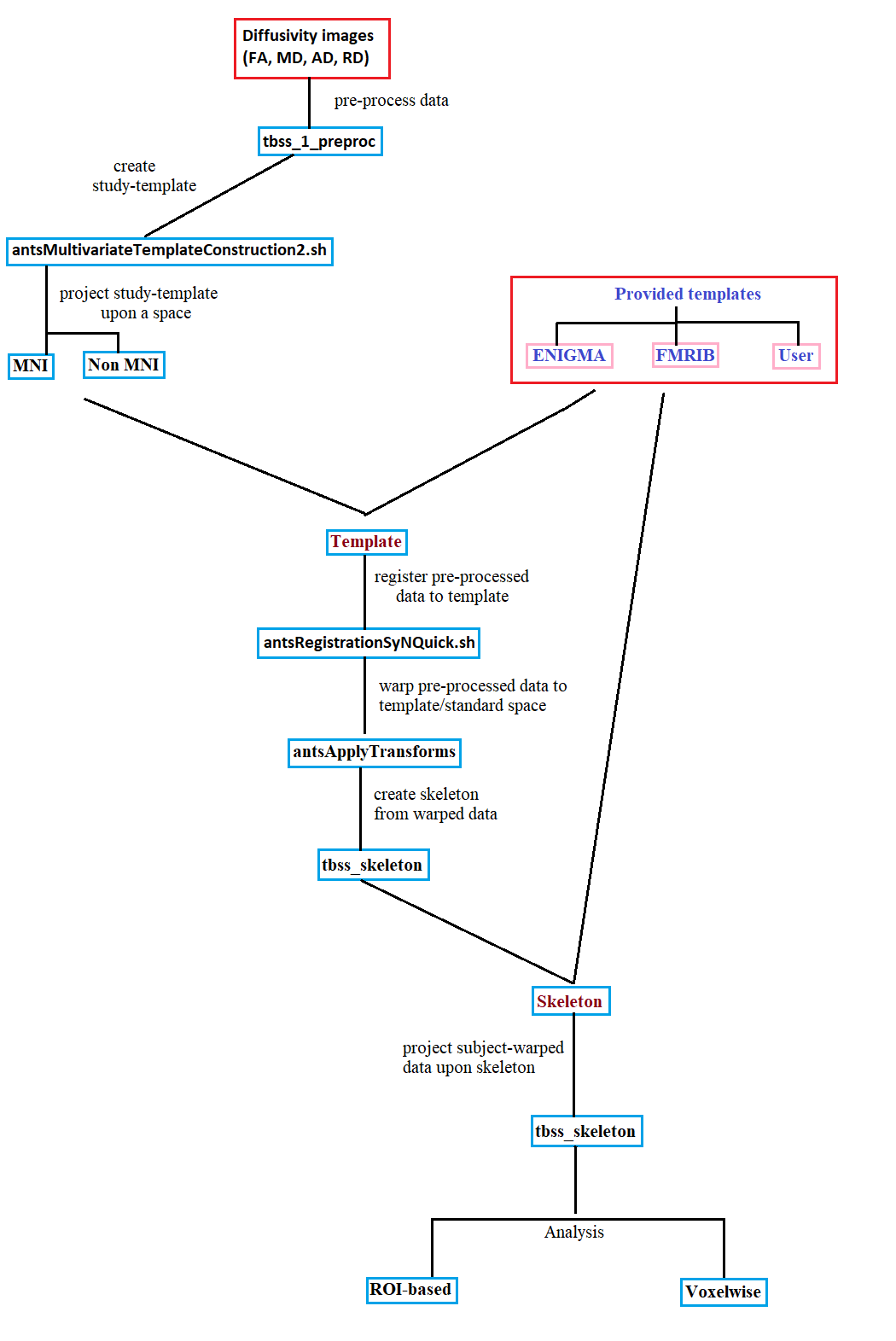
Step-1: Preprocessing
tbss_1_preproc *.nii.gz pre-processes the FA images. It essentially zeros the end slices and erodes the image a little bit.
It also creates caseid_FA_mask.nii.gz that can be used to pre-process non-FA images. tbss_1_preproc puts the given FA
images in origdata and pre-processed FA images in FA directory. The pipeline renames the latter to preproc.
As explained above, non-FA images are pre-processed by applying caseid_FA_mask.nii.gz directly.
Step-2: Registration
Each caseid_FA.nii.gz are registered to template space. The caseid_*1Warp.nii.gz and caseid_*0GenericAffine.mat
transform files are stored in transform/template directory.
The warp and affine are used to warp caseid_FA.nii.gz to template space: caseid_FA_to_target.nii.gz. The warped images
are saved in warped directory. Same warp and affine are used to warp non-FA images.
NOTE If you re-run a study and provided that registration files exist, they will not be re-created. To re-create them,
use --force flag or delete transform/template directory manually. This bypassing would be useful for
quick re-run after adjusting few parameters such as args.SKEL_THRESH.
Step-3: Skeleton creation
If a skeleton is not provided, it is created by tbss_skeleton command. stats/meanFA.nii.gz is used to create skeleton.
The stats/meanFA.nii.gz is obtained from all the warped images in warped directory.
Step-4: Projection
Each subject diffusivity image is projected upon provided/created skeleton: {modality}/skeleton/caseid_{modality}_to_target_skel.nii.gz.
See tbss_skeleton --help for more details about how FA and non-FA images are projected upon skeleton. Also, read Smith’s
TBSS 2006 paper to know more about it.
Step-5: View images
TBSS pipeline generates an HTML file with skeleton overlaid upon the diffusivity measure for all cases.
A slicesdir folder is created inside each modality directory in tbss/output/directory. nilearn package is used to
save .png files with skeleton overlaid upon diffusivity measure for all cases in tbss/output/directory/log/caselist.txt.
Finally, slicesdir/summary.html file is generated with reference to all .png files. Looking at slicesdir/summary.html,
user should be able to assess quality of registration, projection etc. and potentially re-run the pipeline omitting bad cases.
Step-6: ROI/Voxelwise analysis
Finally, we would like to do analysis on skeletonized data. ROI-based analysis can be done as noted in the ENIGMA protocol.
In brief, each caseid_FA_to_target_skel.nii.gz is compared against an atlas. The atlas has multiple segments. We calculate
average diffusivity (FA,MD etc.) of each segment and note them in a csv file: {modality}/roi/caseid_{modality}_roi*.csv.
Summary of ROI analysis is saved in stats/{modality}_combined_roi*csv. The process is detailed in ROI analysis.
On the other hand, skeletonized 4D data stats/all{modality}_skeletonized.nii.gz can be used to do voxelwise analysis.
Branches/Templates
The pipeline has four branches:
1. –enigma
ENIGMA provided templates are used with this argument:
enigmaDir= pjoin(LIBDIR, 'data', 'enigmaDTI')
args.template = pjoin(enigmaDir, 'ENIGMA_DTI_FA.nii.gz')
args.templateMask = pjoin(enigmaDir, 'ENIGMA_DTI_FA_mask.nii.gz')
args.skeleton = pjoin(enigmaDir, 'ENIGMA_DTI_FA_skeleton.nii.gz')
args.skeletonMask = pjoin(enigmaDir, 'ENIGMA_DTI_FA_skeleton_mask.nii.gz')
args.skeletonMaskDst = pjoin(enigmaDir, 'ENIGMA_DTI_FA_skeleton_mask_dst.nii.gz')
args.lut = pjoin(enigmaDir, 'ENIGMA_look_up_table.txt')
In addition, the following atlas is used for ROI based analysis:
args.labelMap = pjoin(fslDataDir, 'atlases', 'JHU', 'JHU-ICBM-labels-1mm.nii.gz')
2. –fmrib
FSL provided templates are used with this argument:
args.template= pjoin(fslDataDir, 'standard', 'FMRIB58_FA_1mm.nii.gz')
args.skeleton= pjoin(fslDataDir, 'standard', 'FMRIB58_FA-skeleton_1mm.nii.gz')
On the other hand, this branch does not do ROI based analysis by default. If wanted, the user should specify an atlas, corresponding space (if atlas and templates are in different space), and look-up-table as follows:
--labelMap atlas.nii.gz -lut my_look_up_table.txt --space MNI.nii.gz
Unlike original TBSS approach, we use the ENIGMA approach that identifies the direction of projection onto the skeleton based on the individual FA maps rather than on the mean FA map.
3. –studyTemplate
With this branch, a study-specific template is created using antsMultivariateTemplateConstruction2.sh.
tbss_1_preproc INPUTDIR/*.nii.gz pre-processes the given FA images.
The pre-processed FA images are used in template construction. Again, the use should provide
a set of FA images for study specific template construction.
4. User template
Finally, the user can specify any or all of the following:
--template TEMPLATE an FA image template (i.e ENIGMA, IIT),
if not specified, ANTs template will be created from provided images,
for ANTs template creation, you must provide FA images,
once ANTs template is created, you can run TBSS on
non FA images using that template
--templateMask TEMPLATEMASK mask of the FA template, if not provided, one will be created
--skeleton SKELETON skeleton of the FA template, if not provided, one will be created
--skeletonMask SKELETONMASK mask of the provided skeleton
--skeletonMaskDst SKELETONMASKDST skeleton mask distance map
** NOTE ** Attributes provided as user templates are mutually exclusive to the ones default with branches specified above.
In other words, branch specific templates have precedence over user template.
For example, if --enigma is specified, it will override --template, --skeleton etc specified again.
However, since --fmrib comes with only --template and --skeleton,
you should specify --templateMask, --skeletonMask etc. with it.
Caselist
File names in each subdirectory start with a caseid obtained from --caselist.
There must be one caseid in each line:
case1
case2
case3
...
Input images
The TBSS pipeline requires input images i.e. FA, MD etc. You may specify the input images as a list or as a directory which contains them.
1. With dwi/mask image list
For convenience, TBSS can start by creating diffusivity measures: FA, MD, AD, and RD. To let the pipeline create them, specify your input DWI/Mask in a text file as follows:
-i INPUT.csv a txt/csv file
with dwi1,mask1\ndwi2,mask2\n...
In addition, provide the --generate flag.
NOTE There must be one dwi,mask pair in each line, separated by comma:
dwi1,mask1
dwi2,mask2
dwi3,mask3
...
Then, FA, MD, AD, RD are created using either DIPY/FSL diffusion tensor models. The model and
associated cost function can be specified by environment variables DTIFIT_TOOL and COST_FUNC
respectively:
export DTIFIT_TOOL=FSL # default DIPY
export COST_FUNC=WLS # default LS (least squares/weighted least squares)
Finally, TBSS is done for specified --modality.
2. With diffusivity image list
Alternatively, you can specify a list of diffusivity images sitting in different directories:
a txt/csv file with
-i INPUT.csv ModImg1\nModImg2\n... ; TBSS will be done for specified Modalities
The pipeline will organize them in proper directory structure.
NOTE Let’s say you have two modalities FA and MD and one INPUT.csv. Then, you would use:
-i INPUT.csv --modality FA,MD
where INPUT.csv looks like following:
/path/to/FA/image1,/path/to/MD/image1
/path/to/FA/image2,/path/to/MD/image2
/path/to/FA/image3,/path/to/MD/image3
...
As you see, first column corresponds to FA images, while the comma separated second column corresponds to MD images.
3. With diffusivity image directory
Finally, to be compatible with FSL/ENGIMA protocols, you may organize your diffusivity images in separate directories. Then, you can provide the directories to run TBSS on:
--modality FA,MD,... dir/of/FA/images,dir/of/MD/images,...
NOTE When specifiying multiple modalities at a time, make sure to correspond your directory to the right modality.
Space
Provided or created template can be projected to a standard space. For human brain, it should be projected to MNI space. However, for rat/other brains, it may be some other standard space.
If ROI based analysis is to be done using a White-Matter atlas, the template should be projected to the space of the atlas.
List of outputs
Several files are created down the pipeline. They are organized with following folder hierarchy and naming:
1. Folders
outDir
|
------------------------------------------------------------------------------------------------------
| | | | | | | |
| | | | | | | |
transform template FA MD AD RD log stats
| (same inner file structure as that of FA)
|
----------------------------------------
| | | | |
preproc origdata warped skeleton roi
copy all FA into FA directory
put all preprocessed data into preproc directory
keep all warp/affine in transform directory
output all warped images in warped directory
output all skeletons in skel directory
output ROI based analysis files in roi directory
save all ROI statistics, mean, and combined images
2. Files
The following directories are created inside user specified output directory. Files residing in the nested directories are explained below:
i. FA/MD/AD/RD
TBSS run on one or more specified modalities. The FA, MD, .. directories correspond to the modalities. In each modality directory, there are five sub-directories:
FA
|
|
----------------------------------------
| | | | |
preproc origdata warped skeleton roi
copy all FA into FA directory
put all preprocessed data into preproc directory
keep all warp/affine in transform directory
output all warped images in warped directory
output all skeletons in skel directory
output ROI based analysis files in roi directory
Files in each subdirectory start with a caseid obtained from --caselist.
a. preproc
Contains all tbss_1_preproc processed data.
b. origdata
Contains raw diffusivity data.
In fact tbss_1_preproc categorizes raw and preprocessed data into origdata and FA directories, respectively.
The pipeline renames FA as preproc to be harmonious with the genre of data contained within.
c. warped
Preprocessed data are warped to template/standard space applying warp and affine obtained from registering each subject
to the template. warped directory contains warped data.
d. skeleton
Each subject diffusivity image is projected upon provided/created skeleton. This directory contains projected skeletons in subject space.
e. roi
If you choose to do ROI based analysis providing a --labelMap, then a *_roi.csv file is created for each case containing
region based statistics. Additionally, if you use --avg flag, RIGHT/LEFT regions in the ROIs are averaged. The averaged
statistics are saved in *_roi_avg.csv file.
Several files are created down the pipeline. They are organized with proper folder hierarchy and naming:
outDir
|
-------------------------------------------------------------------------
| | | | | | |
| | | | | | |
transform/template FA MD AD RD log stats
ii. transform/template
If a template is given, input images are registered with the template. On the other hand, if a template is not
given/--studyTemplate branch is specified, a template is created in the pipeline at template directory.
Corresponding transform files: *0GenericAffine.mat and *1Warp.nii.gz are created in transform/template directory.
Moreover, same directory is used to store transform files if a template is further registered to another space (i.e. MNI).
iii. log
ANTs registration logs are stored in this directory for each case starting with a caseid. However, the user can print
all the outputs to stdout by --verbose option. In addition, all commands ran before along with when it was run,
are stored in log/commands.txt file.
iv. stats
As the name suggests, all statistics are saved in this directory. Statistics include mean and combined modality images, csv file containing summary of region based statistics etc.
multi-modality TBSS
Unlike requiring to save nonFA TBSS files with a particular name as directed by some protocol, this pipeline is capable of running multi-modality TBSS with any name for input images. All you have to do is, make sure first modality in the specified modalities is FA and corresponding input images are FA. Rest of the modalities should also correspond to rest of the images.
--modality MODALITY Modality={FA,MD,AD,RD ...} of images to run TBSS on
(i) single modality analysis:
you must run --modality FA first, then you can run for other modalities such as --modality AD
(ii) multi modality analysis:
first modality must be FA, and then the rest i.e --modality FA,MD,AD,RD,...
files from FA TBSS analysis are used in rest of the modalities
-i INPUT, --input INPUT
(i) DWI images and masks:
a txt/csv file with dwi1,mask1\ndwi2,mask2\n... ; TBSS will start by creating FA, MD, AD, and RD;
additionally, use --generate flag
(ii) single modality analysis:
a directory with one particular Modality={FA,MD,AD,RD,...} images, or
a txt/csv file with ModImg1\nModImg2\n...
TBSS will be done for specified Modality
(iii) multi modality analysis:
comma-separated multiple input directories corresponding to the sequence of --modality, or
a txt/csv file with Mod1_Img1,Mod2_Img1,...\nMod1_Img2,Mod2_Img2,...\n... ;
TBSS will be done for FA first, and then for other modalities.
(iv) separate nonFA TBSS: if you wish to
run TBSS for other modalities in future, files created
during FA TBSS will be integrated into the nonFA TBSS.
Provide the same --outDir as that of FA TBSS.
All other directories will be realized relative to --outDir.
On the other hand, provide --input and --modality in the same way
as you would provide for FA TBSS.
However, if you wish to run FA first and then other modalities in future, use option (iv) from above. There, you should
provide the same --outDir as that of FA TBSS. The --outDir has a transform directory containing warp/affine obtained
during registration of subject FA to the template/standard space.
This way, we bypass doing the same non-linear registration once again. In addition, provide the type/branch name (one of
enigma, fmrib, study) of TBSS you want to run. Previous registration files and any templates created during
FA TBSS are realized with respect to --outDir that you provide. Furthermore, log/commands.txt file from previous
output directory is exploited to figure out arguments provided to FA TBSS before. In a nutshell, nonFA TBSS will run
with the same settings of previous FA TBSS.
Here are a few sample commands for running separate nonFA TBSS:
nonFA --engima TBSS
$libDir/tbss_all -i MD/origdata,RD/origdata \
-o $testDir/enigmaTemplateOutput/ \ # transform files are obtained from here
--modality MD,RD --enigma # --enigma tells to use enigma templates
nonFA --studyTemplate TBSS
$libDir/tbss_all -i AD/origdata \
-o $testDir/studyTemplateOutput/ \
--modality AD \
--studyTemplate # --studyTemplate tells to use previously created templates from studyTemplateOutput/stats/*
List creation
1. imagelist
You can easily generate list of your FA images as follows:
cd projectDirectory
ls `pwd`/000????/eddy/FA/*_FA.nii.gz > imagelist.txt
Here, we have a bunch of cases in the project directory whose IDs start with 000 and is followed by
four alphanumeric characters. The directory structure to obtain FA images is 000????/eddy/FA/. Inside the
directory, we have an FA image ending with _FA.nii.gz.
NOTE: pwd is used to obtain absolute path
Similarly, you can generate a list of your dwis,masks as follows:
cd projectDirectory
touch dwi_mask_list.txt
for i in GT_????
do
echo `pwd`/$i/${i}_dwi_xc.nii.gz,`pwd`/$i/${i}_dwi_xc_mask.nii.gz >> dwi_mask_list.txt;
done
In the above example, we have a bunch of cases with IDs GT_???? having separate folders.
The dwis of the cases follow the pattern ID_dwi_xc.nii.gz and corresponding masks follow the pattern
ID_dwi_xc_mask.nii.gz.
In the same way, you can define your file structure and file names to obtain an image/case list.
2. caselist
For just caselist, you can do:
cd projectDirectory
ls 000???? > caselist.txt
Use of ???? is detailed above.
Analysis
1. ROI analysis
--enigma and --fmrib branch of the pipeline performs ROI based analysis as default. The way it works is, each of the
projected skeleton in subject space is superimposed upon a binary label map. The binary label map of each ROI is obtained from
each segment in the specified --labelMap (atlas). The segments are labelled with an integer in the atlas.
Two information from each ROI is obtained: Average{FA/MD/RD/AD} and number of voxels. Such info from all the ROI for each
case is saved in a caseid_roi.csv file. Additionally, if you use --avg flag, RIGHT/LEFT regions in the ROIs are
averaged. The averaged statistics are saved in caseid_roi_avg.csv file.
Again, ROI statistics of all the subjects are summarized in {modality}_combined_roi.csv and
{modality}_combined_roi_avg.csv files in the stats folder.
Other optional arguments for ROI-based analysis are
i. --lut
A look up table for names of each integer labels in the atlas
ii. --space
If you create a study-specific template or provide a template that is not in the same space of the atlas, you must provide a T1/T2/FA image in the space of the atlas so the subject image can be warped to the same space.
2. Voxelwise analysis
You may perform voxelwise statistics on 4D skeletonised FA image all_FA_skeletonised.nii.gz following this instruction.
All 4D data are saved in stats folder.
QC
Another merit of TBSS pipeline is automatic integration of quality checked/registration corrected data.
Each diffusivity image is warped to template space. The pipeline lets the user visually check the quality of registration.
Enable the --qc flag and the program will halt until you are done with QC-ing.
Warped images are in warped directory. Merged 4D data are in stats directory, corresponding seqFile for index of volumes are also there. You may use fsleyes/fslview to scroll through the volumes.
If re-running registration is required for any case, save the re-registered images in warped directory with the same name as before.
Press Enter, and the program will resume with your corrected data.
For re-registration of any subject, output the transform files to a temporary directory:
mkdir /tmp/badRegistration/
antsRegistrationSyNQuick.sh -d 3 \
-f TEMPLATE \
-m FA/preproc/caseid_FA.nii.gz \
-o /tmp/badRegistration/caseid_FA
antsApplyTransforms -d 3 \
-i FA/preproc/caseid_FA.nii.gz \
-o FA/warped/caseid_{FA/MD/AD/RD}_to_target.nii.gz \
-r TEMPLATE \
-t /tmp/badRegistration/caseid_FA1Warp.nii.gz /tmp/badRegistration/caseid_FA0GenericAffine.mat
Finally, if needed, you can copy the transform files in the transform directory.
NOTE Replace all the above directories with absolute paths.
Fill holes
Diffusion measures having holes inside the brain region have been observed to yield slightly different results from tbss_skeleton
command. To circumvent this issue, we have developed a script that clamps the brain region to a minimum of 10e-8.
This step accures some time required in preprocessing. The hole filling is done by default and images in outDir/{modality}/origdata/
folder are the ones after filling holes, NOT the ones you provide. If you don’t want to do it,
use --noFillHole flag.
Resource profile
SyN registration by ANTs and skeletonization by FSL are two resource intensive procedures in TBSS pipeline. The profile of a single-threaded ANTs registration and tbss_skeleton is given below (notice the RES column):
| Time (minutes) |
RAM (GB) |
|
|---|---|---|
antsRegistrationSyNQuick |
15 | 1.5 |
tbss_skeleton |
3 | 0.5 |
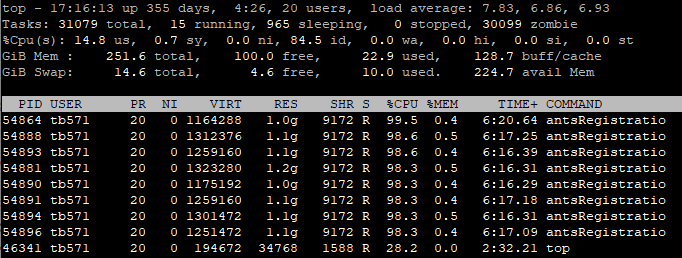
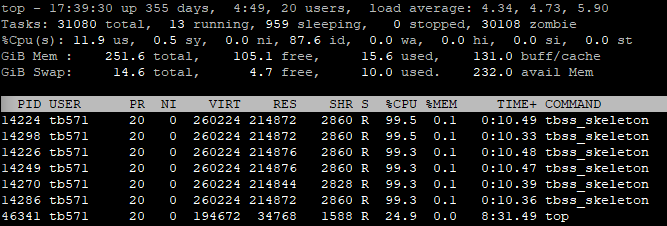
It can be inferred from the above profile that each case is processed in less than 20 minutes consuming less than 1.5 GB of RAM. Of course the actual duration and memory usage would depend on the spatial resolutions of the fixed and moving images– the higher the more.
Multi threading
Processing can be multi-threaded over the cases. Besides, antsMultivariateTemplateConstruction2.sh utilizes
multiple threads to speed-up template construction.
--ncpu 8 # default is 4, use -1 for all available
However, multi-threading comes with a price of slowing down other processes that may be running in your system. So, it is advisable to leave out at least two cores for other processes to run smoothly.
You can check the number of available CPUs using lscpu and available RAM using vmstat -s -S M command.
Then, use a good number of processors permitted by the total RAM such that
each processor can be allocated 1.5 GB of RAM.
For example, if your machine has 16 CPUs and 16 GB RAM, you should use --ncpu 8 which should supposedly
keep RAM usage limited to 12 GB leaving some room for other processes of the machine to continue running.
NRRD support
The pipeline is written for NIFTI image format. However, NRRD support is incorporated through NIFTI –> NRRD conversion on the fly.
See Billah, Tashrif; Bouix, Sylvain, Rathi, Yogesh; Various MRI Conversion Tools, https://github.com/pnlbwh/conversion, 2019, DOI: 10.5281/zenodo.2584003 for more details on the conversion method.
Troubleshooting
(1) The pipeline vastly uses Python multiprocessing. When multiple processes are spawned, RAM is found to be consumed quickly.
Users reported MemoryError which tend to occur when many processors are requested i.e. --npcu 16. The pipeline has
been revised to circumvent such error. Even though you experience one, first step would be to reduce --ncpu. The default
is 4, which most research computers are capable of affording. In any case, it might be useful to reduce it to 2 only.
If it still happens, feel free to open an issue on here.
(2) all_{modality}_skeleton image is computed to facilitate running FSL randomise. When there are many cases (i.e. above 500), merging
the skeletons can be exhaustive in terms of memory. Hence, the program may run into MemoryError. This MemoryError is
distinct from multiprocessing MemoryError. The solution would be to omit merging of skeletons by --noAllSkeleton flag.
(3) A few sub directories are created in your output directory. If the pipeline fails, go to your output directory and find out which subdirectory has less number of images than the cases. You can use a command like below to know that:
cd outDir/FA/preproc
ls | wc -l
If the anomaly is in the first step i.e. preprocessing, two problems might have happened:
(i) Your caselist does not uniquely represent the diffusion measures i.e. one caseied occurs in multiple file names or file corresponding to your caseid does not exist. See 2. caselist to revise the proper way of creating caselist.
(ii) Your image is not readable by FSL. A easy way to find out is by the following:
fslmaths origdata/001.nii.gz -mul 1 /tmp/001.nii.gz
If the above fails, you should recreate your nifti files and make sure they are readable by your version of FSL. FSl version can be checked by
flirt -version
This incompatibility was frequently overserved with FreeWater and FreeWaterCorrected diffusion measures.
(iii) If number of cases/images anomaly exists in other directories, check if your antsApplyTransforms and tbss_skeleton
executables are functioning properly.
(4) See the outDir/log/commands.txt file for history of commands you tried. If you eventually fail to sort out your problems,
open an issue here.
(5) Consider the following inputs:
--input /dir/containing/FA/images/ --caselist caselist.txt
cat caselist.txt
1
11
23
ls /dir/containing/FA/images/
Your_1_FA.nii.gz
My_11_FA.nii.gz
His_23_FA.nii.gz
In the above example, caseid 1 exists as a substring in caseid 11. Hence, the program won’t know whether Your_1_FA.nii.gz or
My_11_FA.nii.gz image is pertinent to caseid 1. In this case, it will raise an error message and ask you to provide explicit
imagelist containing paths as input.
One (or some) of the caseids don't uniquely represent input images.
Either remove conflicting imgs/cases or provide --input IMAGELIST.csv
So, the right inputs would be:
--input imagelist.csv --caselist caselist.txt
cat imagelist.csv
/dir/containing/FA/images/Your_1_FA.nii.gz
/dir/containing/FA/images/My_11_FA.nii.gz
/dir/containing/FA/images/His_23_FA.nii.gz
From the order of files listed in imagelist.csv, now the program knows 1 is associated with /dir/containing/FA/images/Your_1_FA.nii.gz,
/dir/containing/FA/images/My_11_FA.nii.gz is associated with 11 and so on.
(6) If you get any X forwarding error while writing summary.html file containing screenshots of axial, lateral, and sagital views,
omit this functionality with --noHtml flag.
(7) If you re-run a study and provided that registration files exist, they will not be re-created. To re-create them,
use --force flag or delete transform/template directory manually. This bypassing would be useful for
quick re-run after adjusting few parameters such as args.SKEL_THRESH. For adjustments to take effect,
you should run FA TBSS first which will update the parameters in log directory from where
they propagate to subsequent non FA TBSS (altogether or separate).
For --study branch, note that template/template0.nii.gz file may be created even if template creation fails midway.
So, before trying again, make sure to delete that or use --force flag.
(8) log/commands.txt stores all the command you attempt on --outDir. When nonFA TBSS is run separately,
the latest command is parsed
to figure out the following attributes that are same for all modalities:
for i, entity in enumerate(lastArgs):
if entity=='--caselist':
args.caselist= lastArgs[i+1]
elif entity=='--space':
args.space= lastArgs[i+1]
elif entity=='--labelMap':
args.labelMap= lastArgs[i+1]
elif entity=='--lut':
args.lut= lastArgs[i+1]
elif entity=='--SKEL_THRESH':
args.SKEL_THRESH= lastArgs[i+1]
elif entity=='--SEARCH_RULE_MASK':
args.SEARCH_RULE_MASK= lastArgs[i+1]
elif entity=='--avg':
args.avg= True
elif entity=='--noFillHole':
args.noFillHole= True
elif entity=='--noAllSkeleton':
args.noAllSkeleton= True
elif entity=='--noHtml':
args.noHtml= True
If you have made a ‘blatant’ mistake in your attempt of nonFA TBSS, it might be useful to delete the last line
from log/commands.txt lest that line should become the reference command for future nonFA TBSS.
Reference
S.M. Smith, M. Jenkinson, H. Johansen-Berg, D. Rueckert, T.E. Nichols, C.E. Mackay, K.E. Watkins, O. Ciccarelli, M.Z. Cader, P.M. Matthews, and T.E.J. Behrens. Tract-based spatial statistics: Voxelwise analysis of multi-subject diffusion data. NeuroImage, 31:1487-1505
E. Garyfallidis, M. Brett, B. Amirbekian, A. Rokem, S. Van Der Walt, M. Descoteaux, I. Nimmo-Smith and DIPY contributors, “DIPY, a library for the analysis of diffusion MRI data”, Frontiers in Neuroinformatics, vol. 8, p. 8, Frontiers, 2014.
Billah, Tashrif; Bouix, Sylvain, Rathi, Yogesh; Various MRI Conversion Tools, https://github.com/pnlbwh/conversion, 2019, DOI: 10.5281/zenodo.2584003.